
 Open Access
Open Access Abstract
Background: Malaria infection is a multisystem pathology with various clinical complications in both adults and children. The clinical manifestation originates in humans following the invasion of erythrocytes by merozoites.
Methods: The relevant information and data was collated from scientific databases such as Google Scholar, Science Direct, PubMed, Mendeley, Springer Link, and Medline using keywords such as ‘severe malaria infection’, ‘pathophysiology of severe malaria’, ‘complications of severe malaria’ and ‘erythrocyte impairment in severe malaria’.
Results: Generally speaking, the pathophysiology of severe malaria infection encompasses a succession of stages involving the metabolic products of the malaria parasites inclusive of hemoglobin digestion, damaged erythrocyte membrane components, the actions of the pro- and anti-inflammatory cytokines, and the cytoadherence of the malaria parasites to the vascular endothelium as well as sequestration and rosetting. The major complications connected with severe malaria infection include acute respiratory distress syndrome, neurological disorders resulting from cerebral malaria, liver and kidney dysfunction, anaemia and thrombocytopenia, and fatal placental malaria.
Conclusion: The effective management of severe malaria infection involves a proper diagnosis followed by the subjection of the patient to suitable antimalarial treatment with the necessary medications depending on the various clinical manifestations of the infection.
Introduction
Malaria is one of the more widespread health issues worldwide 1 caused by the obligate intra-erythrocytic protozoa of the genus Plasmodium , of the following species, specifically P. falciparum , P. vivax , P. ovale , and P. malariae and in addition, recently P. knowlesi infecting humans 2 . It is noteworthy to mention that the most severe complications and deaths connected with malaria are caused by the P. falciparum species . Other species, namely P. malariae , P. vivax, and P. ovale, rarely induce pernicious complications, debilitating relapses, or death 3 . The incidence of severe malaria is about 2 million cases annually, with approximately 430,000 deaths in the same time period 4 . A total of 435,000 deaths were reported as a result of malaria globally in 2017, of which 80% of the deaths occurred in Africa. Children under 5 years old are most susceptible to the malaria infection, accounting for 61% of the total deaths from malaria in 2017 5 . Malaria is an emergency pathologic condition that is accompanied by a multisystem disorder with various clinical complications in adults and children 6 , 7 , 8 .
Malaria is a life-threatening disease whose clinical manifestation originates in humans following the invasion of erythrocytes by merozoites. After the development of the parasite within the erythrocyte, various waste substances, including the hemozoin pigment and other toxins, accumulate in the infected erythrocyte. These substances are transferred into the vascular system following the lysis of the infected erythrocyte alongside the discharge of the invasive merozoites. There is also the activation of the macrophages and other cells by hemozoin and other malaria parasite toxins. This eventually leads to the generation of cytokines and other soluble factors that play a major role in the initiation of a fever and other pathologies connected with severe malaria 9 . By implication, the pathophysiology of severe malaria infection encompasses a succession of stages involving the metabolic products of the malaria parasites following hemoglobin digestion, damaged erythrocyte membrane components, the actions of the pro- and anti-inflammatory cytokines, and the cytoadherence of the malaria parasites to the vascular endothelium as well as sequestration and rosetting.
Most complications connected to P. falciparum infections are induced by severe anemia or cerebral malaria. However, varying clinical symptoms also occur based on the parasite species and the organ affected 10 . Furthermore, the parasite and host-related factors play a major role in the origin and development of severe malaria infections. Infected erythrocytes adhere to the vascular endothelium of various organs, forming rosettes, thereby initiating vascular damage and a host inflammatory/immune response. The differing presentations of thrombocytopenia, acute respiratory distress syndrome (ARDS), and renal and hepatic impairment, including fatal placental malaria, are other complications connected with severe malaria infection 10 , 11 , 12 , 13 . The present review summarizes the pathophysiology of severe malaria and the varying complications connected with the disease.
Methods
The relevant information and data was collated from scientific databases such as Google Scholar, Science Direct, PubMed, Mendeley, Springer Link, and Medline using keywords such as ‘severe malaria infection’, ‘pathophysiology of severe malaria’, ‘complications of severe malaria’ and ‘erythrocyte impairment in severe malaria’.
Results
Scholarly publications from 1982 - 2020 were chosen from the scientific search engines, resulting in a total of 157 references that have been cited in this review work.
Pathophysiological Processes Involved in the Development of Severe Malaria
Physiological impairment induced by malaria parasites, especially P. falciparum , result in alterations to the normal structure and function of the erythrocytes. This eventually initiates life-threatening complications. In addition, non- falciparum infections such as those by P. vivax and P. knowlesi are involved in this pathology and they have been reported to cause severe complications and death in individuals 14 , 15 , 16 , 17 , 18 , 19 , 20 .
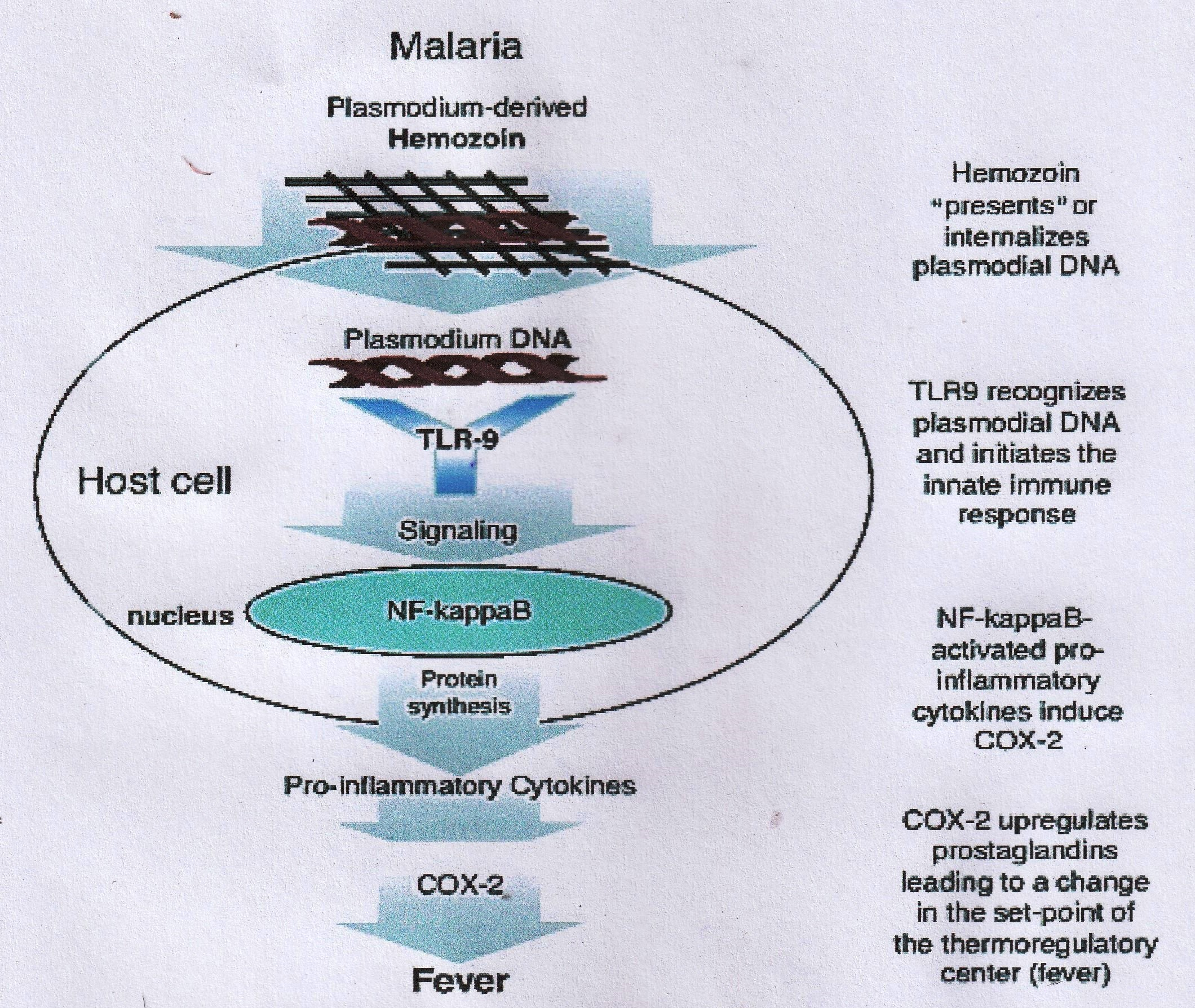
Generally, the pathophysiological events involved are as follows: When the malaria parasites have completed schizogony in the erythrocytes (usually within 24 - 72 hours), the lysis of the infected erythrocytes occurs, leading to the discharge of the merozoite by-products including the damaged erythrocyte membrane components such as glycosylphosphatidylinositol (GPI) and digested hemoglobin– hemozoin pigment. These by-products, especially the GPI and hemozoin, induce the macrophages and endothelial cells to release cytokines and inflammatory mediators like interleukins (IL), namely, IL-1, IL-6, IL-8, lymphotoxin, interferon-γ (IFN-γ), nitric oxide, and superoxide 21 , 22 . The cytokines and membrane products released during the process of erythrocyte lysis have been reported to be accountable for most of the complications that are connected with malaria, such as a fever, headache, weakness, pain in the muscles and joints, diarrhea, central nervous system disorders, stomach discomfort, vomiting, a low blood platelet level, blood clotting impairments, the suppression of the immune system, etc . ( Figure 1 ) 23 , 24 . Similarly, the DNA of the Plasmodium species is a highly pro-inflammatory agent that can initiate fever and the release of cytokines into the bloodstream. The DNA of Plasmodium species internalized by hemozoin (which is formed in the developmental stage of the parasite in the erythrocytes) undergoes an intracellular interaction with Toll-like receptor-9. This elicits the liberation of pro-inflammatory cytokines and the activation of the cyclooxygenase (COX)-2-mediated upregulation of prostaglandins biosynthesis responsible for the initiation of a fever ( Figure 2 ) 25 , 26 . Hemozoin has also been reported to cause anemia by enhancing the extermination of developing erythrocytes in the bone marrow 27 , 28 .
Figure 1 . Sequence of events leading to severe malarial infection (Source 23 ).

Figure 2 . Initiation of fever by the plasmodial DNA (Source: 26 ).
 26
)." width="300" height="200">
26
)." width="300" height="200">
Role of Cytokines in Severe Malaria
The cytokines as part of the pro-inflammatory cascade such as IFN-γ, ILs, tumor necrosis factor (TNF) and nitric oxide play a foremost role in initiating malaria complications. However, the low plasma levels of cytokines have been reported to ameliorate the virulence of malaria parasites. The failure of the cellular down-regulatory mechanisms in the early stages of the pro-inflammatory cytokine response is responsible for the impairment of the immune system, leading to various connected disorders to severe malaria. Very high cytokine levels have been reported to elicit the following pathophysiology, namely a reduction in the utilization of mitochondrial oxygen, the stimulation of lactate production, the induction of microvascular obstruction, low oxygen levels enhanced by cytoadherence, the obstruction of blood circulation, dyserythropoietic and multifactorial anemia, the inhibition of gluconeogenesis, low glucose levels and myocardial impairment. Additionally, elevated levels of cytokines in the plasma enhance the pro-coagulation process through the stimulation of the leukocytes or platelets, in addition to endothelial injuries as well as vascular damage in the brain and lungs and the upregulation of the cranial and placental vascular and intercellular adhesion molecules (ICAMs) that results in cerebral malaria and drastically affects placental functions 21 , 22 , 26 , 29 , 30 , 31 , 32 , 33 , 34 , 35 . The equilibrium between pro- and anti-inflammatory cytokines determines the manifestation and severity of the malaria infection 22 , 30 , 32 .
Cytokine-mediated injury has also been reported to be connected to the complications encountered in P. vivax malaria. P. vivax is known to initiate a higher inflammatory response than P. falciparum due to the greater release of cytokines. P. vivax infections are depicted by a lower pyrogenic threshold, meaning that they induce a fever at lower parasitaemia levels. The greater pyrogenicity of P. vivax has been attributed to its structural differences in terms of glycosylphosphatidylinositol and higher levels of Toll-like receptor-9-stimulating motifs in its hemozoin pigment 18 , 36 .
Figure 3 . Cytoadherence, sequestration and resetting of P. falciparum infected erythrocytes (Modified from 37 ).

Cytoadherence and Sequestration
Cytoadherence refers to the efficiency of the parasites to bind to the vascular endothelium 10 . Mature parasites in the asexual stage and the gametocytes can cling to the vascular endothelium of various organs, including the heart, liver, lung, kidney, and brain, in addition to the placenta and subcutaneous adipose tissues ( Figure 3 ) 11 , 12 .
Sequestration refers to the process whereby infected erythrocytes adhere to the endothelial cells of the capillaries and venules, usually in the late stages of the development of the parasite (trophozoites and schizonts) ( Figure 3 ) 38 . The sequestration of parasites within the endothelial cells is, for the most part, the pathological basis of severe malaria infection with cerebral malaria inclusive of this 39 . Parasite sequestration induces local hypoxia through the obstruction of normal blood circulation. Additionally, malaria parasite sequestration stimulates the replication of the parasite and the adhesion of parasitized erythrocytes to the non-infected erythrocytes (a process known as rosetting). Rosetting increases the rigidity of both the normal and infected erythrocytes 40 . Consequently, malaria parasite sequestration causes the localization of tissue damage that is incurred by the parasite toxins and the activation of the host immune response. This can possibly initiate a focused inflammatory mediator and functional tissue impairment.
Matured forms of the malaria parasites are mostly involved in the sequestration process. This occurs about 20 hours after the erythrocyte invasion. New proteins are synthesized by the parasite which are conveyed to the parasitized erythrocytes' surface, enhancing the infected erythrocyte adhesiveness to the endothelium. There is the possibility of the malaria parasites remaining sequestered in the deep microvasculature for 24 hours during their 48-hour lifecycle. This enables the malaria parasites to elude clearance by the spleen, complicating diagnosis because they can no longer be sighted in the peripheral blood 10 .
Certain molecules that originate from the parasite and ligands found on the human endothelium have been reported to play a foremost role in the sequestration process of P. falciparum . The major molecules identified to be responsible for this effect include the P. falciparum histidine-rich protein (PfHRP) and the P. falciparum erythrocyte membrane protein 1 (PfEMP1). PfHRP initiates the generation of knobs (symmetric membrane arrangement) on the infected erythrocyte surface. At the same time, PfEMP1 pops out from the knobs and contributes extensively to the sequestration and virulence of the parasite 11 , 12 . The adherence of the parasite to the endothelium occurs in the following order of events. The parasite first adheres loosely to the endothelium, followed by rolling. The parasite then strongly attaches itself to the adhesion molecules of the endothelium ( Figure 3 ) . During this process, the sequestration receptor designated as intercellular adhesion molecule-1 (ICAM-1) functions as the rolling receptor, while CD36 ensures stationary and stable adherence under flow 41 , 42 .
Sequestration also occurs in gestational malaria, during which the parasite binds to the placenta. PfEMP1, which is still the major adhesion receptor, binds to the trophoblastic villous endothelium via the chondroitin-4-sulfate and glycosaminoglycans and probably hyaluronic acid (HA). Severe forms of malaria may occur in pregnant women which may lead to fetal death, particularly among women undergoing their first pregnancy. This is because these women have not developed enough of an immunity against the chondroitin-4-sulfate binding parasites 43 , 44 , 45 .
Rosetting
Rosetting is a process that involves the cytoadherence of the late stages of an infected erythrocyte to uninfected/parasitized red blood cells (PRBCs) and/or thrombocytes, leading to the formation of erythrocyte rosettes ( Figure 3 ) 46 . PfEMP1 is the infected erythrocyte ligand that plays a major role in the generation of the rosette. Rosetting takes place through the adhesion of the lectin-like DBL-domain of PfEMP1 present on the infected erythrocyte surface to the complement receptor 1, CD31, and to the heparin sulfate-like glycosaminoglycans of uninfected erythrocytes 47 , 48 , 49 , 50 . The rate of rosette formation in blood Group O erythrocytes is much less when compared to blood groups A, B, and AB erythrocytes. Individuals that possess blood group O erythrocytes may not suffer from the severe form of malaria in this regard 50 , 51 .
Although P. falciparum , P. vivax , and P. ovale have all been reported to be involved in the formation of rosettes, only the rosettes formed by P. falciparum have the capability to initiate severe malaria 10 , 52 . The biomembrane lipid peroxidation product– 4-hydroxynonenal (4-HNE) is probably transported from the infected erythrocyte to the normal erythrocyte in rosettes, thereby enhancing the elimination of the normal erythrocytes by macrophages. This is possibly the cause of the high rate of loss of normal uninfected erythrocytes during severe malarial anemia 53 .
Rigidity and Deformability of the Erythrocyte Membrane
Distortion in the rigidity and deformability of the erythrocyte membrane plays a foremost role in the emergence of severe malaria complications. In the presence of severe falciparum malaria, both the PRBCs and non-PRBCs become rigid 37 , 54 , 55 . An increase in the erythrocyte membrane rigidity and a decrease in membrane deformability arises due to the oxidative damage of the erythrocyte membrane engendered by hemin, distortions in the membrane phospholipid bilayer and the attached spectrin network, and thermally driven membrane fluctuations caused by pyrexia, including the blockage of the Na + /K + pump on erythrocyte membrane. This is caused by nitric oxide 22 , 55 , 56 .
A decrease in erythrocyte membrane deformability causes a rise in splenic clearance and a loss of erythrocytes, which leads to anemia. In as much as there is a decrease in erythrocyte membrane deformability in P. falciparum infection, there is an increase in erythrocyte membrane deformability in P. vivax infection. Although an increase in erythrocyte membrane deformability may enable P. vivax infected erythrocytes to bypass the splenic sinusoids without being destroyed, the increased fragility of the erythrocytes as a result of the increased erythrocyte deformability can also induce severe anemia in P. vivax malaria, as well as inhibiting sequestration and enabling the free flow of blood 18 , 57 .
Complications Associated with Severe Malaria Infection
The impairment of the erythrocyte physiochemistry initiates the complications connected with malaria infection through the asexual forms of the malaria parasite. Malaria is a potentially multisystem disease because the erythrocytes that are negatively affected during the malaria infections are needed by all of the organs in the body system 32 , 58 . Fever is a major known symptom of malaria infection, although the illness can advance into the severe malaria form. Even though P. falciparum is the foremost initiator of severe malaria, deleterious health effects and some death cases have been reported to be connected with non- falciparum malaria 5 .
Anaemia
Anemia has been labeled the highest cause of morbidity and death in relation to the malaria infection, especially in children and pregnant women 10 , 59 . Malaria anemia may occur in either an acute or chronic state. The chronic form of malaria anemia is rampant in holo-endemic areas. Acute malaria anemia emanates from the lysis of the erythrocytes caused by increased parasitemia or drug-induced/immune hemolysis 60 .
The pathophysiological events that result in malaria anemia can be categorized into two groups: the increased damage of PRBCs and non-PRBCs by immune-mediated lysis, phagocytosis, and splenic sequestration in addition to a reduced rate of erythrocyte formation as a result of dyserythropoietic and secondly, the suppression of the bone marrow, the reduced formation of reticulocyte, the actions elicited by inflammatory cytokines and other parasite factors. Furthermore, the reduction in hemoglobin level in malaria patients is induced by co-infection with bacteria, hookworm, and human immune-deficiency virus-1 (HIV-1), poor nutrition, and reoccurring malaria infections in holo-endemic countries 59 , 60 , 61 .
The major identified causes of malaria anemia are the increased propensity of PRBCs to rupture and the rapid splenic clearance of distorted PRBCs and non-PRBCs. Non-PRBCs can be prematurely eliminated from the blood by mechanisms involving the rigidity of the membrane, phospholipid asymmetry, and decreased deformability, as previously described 40 . The action of the spleen has been reported to be a contributory factor in severe malaria by virtue of its capacity to remove PRBCs and non-PRBCs from the systemic circulation excessively. Nevertheless, the spleen can offer protection against severe cerebral malaria 62 , 63 .
Dyserythropoiesis is a major contributor to the pathogenesis of anemia. The phagocytosis of hemozoin by the bone marrow macrophages is known to induce dyserythropoiesis 62 . Additionally, the immune response plays a principal role in malaria anemia pathogenesis. The monocyte and lymphocyte responses are known to be stimulated by PRBCs, malaria antigens, and hemozoin. In addition, the pro-inflammatory and anti-inflammatory mediators such as IFN-γ, IL-1, IL-23, TNF-α, chemokines, and growth factors generated initiate anaemia 60 , 64 . The macrophage migration inhibiting factor (MIF) is another contributor to severe anemia in malaria infection due to the MIF-induced suppression of bone marrow erythropoiesis 65 .
Pro-inflammatory cytokines are involved in the iron delocalization pathway of anemia in severe malaria. Ferroportin delocalization is activated by TNF-α. Ferroportin is an essential protein that is found in large amounts in the reticuloendothelial system which mediates the macrophage iron discharge and the absorption of iron in the intestine. Ferroportin delocalization promotes a reduction in iron absorption and macrophage cell release 66 . Hepcidin is known to stimulate the decreased level of ferroportin in the reticuloendothelial system, and the level of hepcidin is elevated in severe malaria anaemia 67 .
Micronutrient malnutrition, especially a lack of vitamins A and E, riboflavin, iron, folate, and zinc, has been reported to escalate the complications connected with anemia by disrupting the immune system and iron metabolism dyserythropoiesis 59 .
Thrombocytopenia
Thrombocytopenia is a common disorder encountered in malaria infection, especially in the early stage of the infection. It is induced by P. falciparum and P. vivax 8 , 68 , 69 , 70 . The occurrence is very prevalent in children and adults. According to the study carried out by Tan et al . 71 in Thailand, lower platelet counts were reported in pregnant women in comparison to non-pregnant women. The pathogenetic features of malaria thrombocytopenia are vast and may be related to coagulation impairments, splenomegaly and damage to the platelets by macrophages, in addition to the distortion of the bone marrow, oxidative stress, and aggregation of platelets 72 . Low platelet count and a high level of the von Willebrand factor were reported in P. falciparum and P. vivax malaria-infected individuals in Indonesia. The High von Willebrand factor is known to be related to platelet binding and therefore can elicit thrombocytopenia 73 . Another study by de Mast et al . 68 indicated an elevation in the external domain of the platelet receptors for the von Willebrand factor (sGP1b) in the blood of individuals suffering from malaria, thereby inhibiting increased platelet adhesion.
Malaria-associated Acute Respiratory Distress Syndrome (MA-ARDS)
The most genuine complexities seen in intestinal sickness are metabolic acidosis, pulmonary complications, placental malaria, cerebral malaria, and hemorrhages with serious anemia, all of which can prompt ARDS 74 .
Tragically, there is relatively little epidemiological information on MA-ARDS 75 . It happens for the most part in grown-ups with fast and helpless anticipation with a lethality pace of 20 — 80%, even with antimalarial treatment 76 . Manifestations range from gentle respiratory inconveniences, for example, dyspnea and a cough, to an advancement in ARDS 75 , 76 .
In MA-ARDS, most detailed cases are in a low transmission zone or are experienced by non-insusceptible voyagers 77 . ARDS is connected with intense irritation and damage to the alveolar endothelium and aspiratory parenchyma, which subsequently causes dysfunctions and an expanded porosityof the pneumonic alveolar-capillary hindrance. This promotes the development of edema 78 . Diminished gas exchange and expanded inflammatory mediators in the lungs bring about respiratory distress in sick patients, prompting death 77 , 78 , 79 .
The full intricacy of human MA-ARDS has provoked the necessity to comprehend the physiopathology of the malaria infection 76 . Accordingly, the murine models have facilitated the understanding of the various intricacies of MA-ARDS pathogenesis, mainly neutrophil inclusion and the levels of hypoxemia like those seen in humans 76 , 80 , 81 .
The clinical manifestations identified with MA-ARDS resemble those of ARDS caused by different illnesses in addition to the malaria side effects depicted previously. It is expected to begin with less severe intricacies, for example, a cough and dyspnea advancing until the development of pneumonic edema 75 , 82 . Although the MA-ARDS mechanisms are yet not well established, previous research reports have shown the significance of CD8 + T cells in murine MA-ARDS and the activity of leukocytes integrin in the pathogenesis of MA-ARDS 83 , 84 .
Nonetheless, there are two primary events that explain the pathogenesis of MA-ARDS. One is centered on the fiery reaction of the host related to the cellular dysfunction of the pneumonic microvasculature 85 , 86 and the other is identified by the adhesion of the infected erythrocytes to the endothelial cell of the pulmonary endothelium that promotes the pathogenesis of the infection 87 , 88 . In the lung tissue, the exudative stage is in the beginning stage of the condition where harm to the endothelial hindrance happens because of endothelial cell necrosis, bringing about edema that pours out into the alveoli. This causes the development of a hyaline layer in the alveolar wall 80 . The histological segments taken from MA-ARDS patients showed bountiful leukocytes, chiefly macrophages in the tissue or alveolar spaces, and a lower number of lymphocytes and neutrophils 75 , 76 . The proliferative fibro stage happens in a later period and it is related to fibroblast cell multiplication and collagen deposition 76 , 80 .
MA-ARDS is associated with the alveoli damage arising from endothelial and epithelial cells injuries, the increment of vascular porousness, and the intercellular spaces followed by edema. During the intra-erythrocyte cycle, the parasite processes hemoglobin to form a compound of the toxic heme group, hemozoin. This prompts the delivery and initiation of pro-inflammatory elements like chemokines, interferon-ɣ (INF-ɣ), CXC-chemokine ligand-10 (CXCL10), CC-chemokine ligand2 (CCL2), keratinocyte-derived chemokine (CXCL1), TNF, IL-1β, IL-6, IL-8, IL-10, transforming growth factor–β (TGF-β) and other inflammatory mediators, for example, heme oxygenase–1 (HO-1) 75 , 89 , 90 .
In P. falciparum malaria, PRBCs exhibit a high propensity to adhere to endothelial cells of several organs microvasculature due to the expression of PfEMP1 91 . Proteins intervene in the attachment to non-PRBCs and PRBCs to form rosettes that advance the adhesion to few receptors like CD36, ICAM-1, vascular adhesion molecule-1 (VCAM-1), chondroitin sulfate A (CSA)88, and the endothelial protein C receptor (EPCR) 92 present in the endothelial microvasculature of various tissues, for example, cerebral, pneumonic and placental. This attachment permits the parasite to complete its life cycle without being wiped out by hemocatheresis. This exacerbates the severity of the disease 88 . Notwithstanding, it isn't clear which bond is particularly responsible for the interface between the infected erythrocytes and pneumonic vascular endothelium in MA-ARDS 79 , 83 , 90 . Recent research has demonstrated that the development of neutrophil extracellular traps (NETs) is potentially connected to the severity of MA-ARDS and the different intricacies of malaria infection 93 , 94 . Within the blood vessels, NETs shields the endothelial obstruction from the inflammatory components, simultaneously actuating the complement system, influencing hemostasis, designing scattered intravascular coagulation eliciting hemorrhages and thrombi formations, and, subsequently, ischemia 94 . In vivo and in vitro experiments performed by Sercundes et al ., utilizing murine models showed that the treatment needed to restrain the NETs developments resulted in a critical improvement in the MA-ARDS pathology 93 .
Neurological Complications (Cerebral Malaria)
A major clinical manifestation, which in certain cases leads to death in severe malaria among adults, is cerebral malaria. In the early stage, it is characterized typically by generalized convulsions or drowsiness and confusion, eventually leading to a coma 95 . From an experimental point of view, changes in the normal mental status of an individual should be handled as though it is cerebral malaria. Furthermore, delirium, agitation, and transient paranoid psychosis can also occur when the patient regains consciousness. Other neurological sequelae different from cerebral malaria, which can also happen in severe malaria, include cranial nerve abnormalities, ataxia, and extrapyramidal tremor 2 . About 10-50% of the total cerebral malaria cases have been reported to lead to death, even when under treatment 96 .
Mild stiffness of the neck may occur in this case. Retinal hemorrhages have been observed in about 15% of the total cases investigated. Other common major complications of this condition include fixed jaw closure and tooth grinding (bruxism) 5 .
Nevertheless, varying disorders such as a dysconjugate gaze, pouting, decerebrate and decorticate rigidity, opisthotonus, vessel changes, plantar reflexes and deep jerks, and papilledema, anemia, hepatosplenomegaly, retinal whitening, and jaundice may possibly occur 5 , 97 , 98 .
Liver Pathology in Severe Malaria Infection
The liver is a principal organ that is required during the hepatic phase of the life cycle of the malaria parasite as it is where malaria sporozoites transform into merozoites. The merozoites are delivered into the systemic circulation and enter the erythrocytic stage. In the erythrocytic stage, PRBCs become sequestered in the small blood vessels. The debased haemozoin color is then inundated by nearby tissue macrophages like alveolar and macrophage Kupffer cells. Histopathological observations of the liver in P. falciparum malaria infection incorporate responsive Kupffer cells, the maintenance of the haemozoin color, and insignificant PRBC sequestration 99 , 100 . An ultrastructural study showed there to be a relationship between a high PRBCs load in the liver of malaria patients with jaundice, hepatomegaly and liver enzyme elevation 101 .
Apoptotic changes occur in various cell systems and include both pathological and physiological changes. While the liver has not been reported to exhibit an apoptotic change in human malaria, these changes have been reported in animal models during the erythrocytic stage in hepatocytes and in the hepatic stage of Kupffer cells 102 , 103 , 104 . The process of programmed cell death can intercede in the presence of different enhancers including hormones, growth factors, cytokines, bacterial or viral diseases, and their resistant reactions 105 . Cell apoptosis is regulated through two significant pathways: the natural or mitochondrial pathway and the extraneous or death-receptor pathway. Initiator caspases, for example, caspase-8 or -9, assumes a regulatory part by enacting downstream effector caspases, for example, due to caspase-3, -6, or -7 106 . Nuclear factor-kappa B (NF-κB) has been seen to manage the apoptotic program in different cell types, either as an up-controlling reaction or as an apoptosis blocker 107 . Proof of NF-κB directing apoptosis has been observed in the cerebrum endothelial cells and intravascular lymphocytes in cerebral malaria 108 .
Likewise, hyperbilirubinemia (> 3 mg/dl) and raised plasma transaminases are diagnostic of malarial hepatitis 109 . The adhesion of PRBCs to the endothelial walls of the liver vessels prompts the blockage of the intrahepatic ducts, engendering modifications in the bloodstream and as a result, ischemia. Intricacies like hepatic encephalopathy, multi-organ dysfunction, and impaired protein synthesis follow. Histopathological studies have uncovered hepatocyte necrosis, cholestasis, granulomatous sores, and malaria knobs in severe malaria infection 110 .
As per the World Health Organization (WHO) guidelines, indications of malarial hepatopathy are abnormal in instances of severe malaria infection. Indications of liver dysfunction have been reported in Asia, particularly in India. The majority of these cases are due to falciparum malaria or mixed (both falciparum and vivax ) malaria 111 . Cases with adjusted liver capacity tests and surprisingly fulminant hepatic dysfunction have been reported 112 , 113 . An investigation conducted on Nigerian children noted that the elevated plasma levels of liver biomarkers are the biochemical indicators of intense P. falciparum parasitaemia 114 .
Kidney Pathology in Severe Malaria Infection
Malaria parasites were the main parasitic agents to be plainly connected with glomerular infections in tropical zones. Serious malaria can cause pathology of the glomeruli, tubules, and interstitial locale 115 . Kidney infection in malaria is principal because of erythrocyte anomalies. PRBCs will, in general, cling to the capillary endothelium, healthy erythrocytes, and blood platelets, prompting the development of rosettes and clusters, which hinders microcirculation 116 . These pathological events are plausible contributing factors to kidney injury as part of a relationship with hemodynamic instability, including hypovolemia and shock. Endothelial interactions prompt the release of a few cytokines, including thromboxane, catecholamine, endothelin, and other inflammatory mediators that are likewise ensnared in the pathogenesis of malaria-related kidney injury. Invulnerable system actuations in malaria can go through T-cell helper type 1 (Th1) and T-cell helper type 2 (Th2) responses. When the Th2 response prevails in the malaria infection by P. malariae , complement actuation prompts glomerulonephritis. Hemodynamic unsteadiness occurs because extreme erythrocyte parasitism prompts intense tubular necrosis as observed in P. falciparum infection. At the point when Th1 response prevails, intense interstitial nephritis and intense glomerulonephritis can be observed. Cortical necrosis has additionally been observed in severe malaria infection, describing a more serious kidney injury. For the most part, it is connected with the non-recuperation of renal function, and therefore the advancement of end-stage kidney failure 117 . A few factors add to the event of these complications, namely vasoconstriction, hypovolemia and hemolysis (prompting hemoglobinuria), erythrocyte parasitemia, resistant edifices deposition in glomeruli, microcirculation dysfunction (due to the cytoadherence of parasitized erythrocytes), and rhabdomyolysis (which isn't normal in malaria). Another contributing factor to kidney failure in malaria is hepatic dysfunction with jaundice and hepatomegaly, through which hyperbilirubinemia can prompt cast nephropathy and acute kidney injury (AKI). Liver dysfunction and its intricacies can likewise cause AKI (hepato-renal disorder) 118 , 119 , 120 , 121 .
AKI accounts for Plasmodium species ( P. falciparum, P. vivax, P. malariae and P. ovale ) infections and the patient can deteriorate because of the low hydration and liquid loss brought about by pyrexia spewing, perspiring, and parchedness. Histological examinations have shown that glomerulonephritis, interstitial nephritis, and acute tubular necrosis are evidence of AKI. It is likewise conceivable to discover ongoing kidney dysfunction related to malaria, mostly in the patients experiencing repeated cases of infection 116 , 117 , 122 , 123 .
Placenta Pathology in Severe Malaria Infection
Malaria in pregnancy is a significant worldwide public health challenge. In 2018, the WHO reported 11 million pregnancies with a high malaria burden in sub-Saharan Africa 124 . In endemic areas, the incidence of placental malaria can reach up to 63% in pregnant women, regardless of the malaria infection symptomatology 125 , 126 .
Likewise, malaria infection during pregnancy elicits clinical symptoms such as anemia, aspiratory edema, puerperal sepsis, cerebral malaria, hypoglycemia, which can sometimes result in death. In addition, maternal malaria elicits the abortion of pregnancy, intrauterine growth retardation (IUGR), unexpected labor, a low birth weight (LBW), etc . 127
The WHO revealed in 2018 that there were 228 million cases of maternal malaria, of which 93% of such cases occurred in Africa, followed by Southeast Asia (3.4%) and the Eastern Mediterranean Region (2.1%). The prevalence of maternal malaria has been generally steady since 2014 with 57 cases for each 1000 population 128 .
Placental malaria appears to occur following the Plasmodium evasion of the spleen. The Plasmodium binds to the VAR2-CSA proteins that interact with CSA in the placental intervillous space 44 , 129 . Placental malaria is diagnostic of the presence of parasitized erythrocytes in the intervillous space, invading the maternal monocytes/macrophages 130 . Noticeable inflammatory actions by the monocytes/macrophages cause enormous, persistent inter-villositis characterized by serious placental malaria 131 . The inflammatory reactions in placental malaria restrain a vital mechanistic target of rapamycin (mTOR) signaling 132 . In placental malaria, the autophagy-related genes are downregulated, prompting autophagy dysregulation and in this way, hindered the trans-placental amino acid transport 133 . Likewise, the blockage of mTOR signaling as a result of placental malaria prompts a diminished placental amino acid uptake 134 . Recently, it was observed that placental malaria stimulated the placental expression of inflammasomes which were connected to placental discharge and the development of IL-1β, a supportive inflammatory cytokine that engenders a reduced nutrient transporter expression 132 .
Elevations of pro-inflammatory cytokines, oxidative stress, and apoptosis engender pathological alterations in the placenta 135 , 136 . It has been shown through histopathological alterations that placental malaria exacerbates the danger of toxemia in pregnancy, particularly in primigravidae 137 . Histopathological alterations during placental malaria showed the presence of hemozoin, perivillous fibrin deposition, syncytial hitch development, and decline in the villous surface area 138 . These pathologic changes in the placenta may restrict the exchange of nutrients between the mother and embryo, expanding the hazard of limited fetal development and LBW infants 139 , 140 . Placental malaria diminishes the abundance of megalin and disabled homolog 2 (Dab2) in syncytiotrophoblasts, which might be accompanied by LBW 141 . In Papua, studies showed there to be a relationship between diminished birth weight and placental mitochondrial DNA copy number 142 . The previous report showed that malaria infection during early pregnancy prompted the modification of the vascular structure of the placenta, diminished transport villi volume, and an increased diffusion distance and diffusion vessel surface. This had an impact on both birth weight and gestational length 143 . All things being equal, Plasmodium infection in mid-pregnancy is connected with an increased danger of preterm birth, conceivably because of the progressions in the dysregulation of angiogenesis, metabolism and inflammation 144 .
Maternal HIV infection increases the severity of placental malaria by hindering antibody advancement to the variant surface antigens expressed by malaria-infected erythrocytes while also dysregulating cytokine biosynthesis, and decreasing the defensive IFN-γ reactions 145 . Reports show that the incidence of malaria diminished in the setting of antiretroviral therapies, especially the administration of protease inhibitors 125 . Notwithstanding, a new randomized controlled preliminary survey study of HIV-positive pregnant women showed no decline in placental malaria in the presence of a protease inhibitor which was in contrast with non-nucleoside reverse transcriptase inhibitors 146 . Consequently, antiretrovirals may not assume a defensive role in placental malaria infection.
One investigation showed that infants conceived with placental malaria had higher chances of clinical malaria in their first year of life 147 . Another survey showed that clinical malaria presents earlier on in the life of infants conceived with placental malaria compared to infants conceived without placental malaria 148 . A new report established that there were significantly increased chances of the incidence of clinical malaria in neonates conceived by women with placental malaria 149 . However, a deliberate survey of 14 patients reported a lack of proof when affirming the relationship between malaria in pregnancy and malaria in the earliest stages. There was no obvious proof of vertical transmission 150 , 151 and the result of innate malaria is uncommon, making the survey outcome inconclusive.
Placenta malaria is described using the sequestration of P. falciparum -infected erythrocytes and invasion of the intervillous spaces of the placenta. The placenta becomes dark because of the deposition of the malaria pigment-hemozoin. The parasite densities are a lot higher in the placenta compared with peripheral blood 152 , 153 . The thickening of the placental basement membrane, perivillous fibrinoid stores, and the syncytial knotting results in an adjusted exchange system between the mother and embryo. The diminished capacity of the placental to deliver nutrients to the fetus causes IUGR 154 , 155 .
Additionally, previous studies have shown that an increased susceptibility to infections during pregnancy results in high parasitemia and a heavy invasion of parasite-tainted RBCs in the placental vasculature 44 , 156 , 157 . Besides, it has also been observed that malaria infection is more profound in primigravidae than multigravidae. This protection from malaria infection in multigravidae is because of the improvement of the placental parasite-explicit invulnerability in subsequent pregnancies 44 , 157 .
Conclusions
The major complications connected with severe malaria infection include ARDS, neurological disorders resulting from cerebral malaria, liver and kidney dysfunction, anemia and thrombocytopenia, and fatal placental malaria. However, the effective management of severe malaria infection involves a proper diagnosis followed by the subjection of the patient to suitable antimalarial treatments with the necessary medications depending on the various clinical manifestations of the infection.
Abbreviations
4-HNE : 4-Hydroxynonenal
AKI : Acute kidney injury
ARDS : Acute respiratory distress syndrome
CCL2 : CC-chemokine ligand2
COX : Cyclooxygenase
CSA : Chondroitin sulfate A
CXCL1 : Keratinocyte-derived chemokine
CXCL10 : CXC-chemokine ligand-10
Dab2 : Disabled homolog 2
EPCR : Endothelial protein C receptor
GPI : Glycosylphosphatidylinositol
HA : Hyaluronic acid
HIV : Human immune-deficiency virus
HO-1 : Heme oxygenase–1
ICAMs : Intercellular adhesion molecules
ICAM-1 : Intercellular adhesion molecule-1
IL : Interleukin
IL-1β : Interleukin-1β
IFN-γ : Interferon-ɣ
IUGR : Intra-uterine growth retardation
LBW : Low birth weight
MA-ARDS : Malaria-associated acute respiratory distress syndrome
mTOR : Mechanistic target of rapamycin
MIF : Macrophage migration inhibiting factor
NF-κB : Nuclear factor-kappa B
NETs : Neutrophil extracellular traps
PfEMP1 : Plasmodium falciparum erythrocyte membrane protein 1
PfHRP : Plasmodium falciparum histidine-rich protein
PRBCs : Parasitized red blood cells
TGF-β : Transforming growth factor– β
Th1 : T-cell helper type 1
Th2 : T-cell helper type 2
TNF : Tumoral necrosis factor
VCAM-1 : Vascular adhesion molecule-1
WHO : World Health Organization
Acknowledgments
Not applicable.
Author’s contributions
FOO/PCC conceived and designed the scope of the report. FOO/PCC/CMC/CCA/FCL-O contributed in writing the paper. FOO/PCC revised and edited the manuscript draft. CMC/CCA/FCL-O authors were the resource persons who provided all the necessary materials for writing the manuscript. All authors have read and approved the manuscript in the present form and gave the permission to submit the manuscript for publication.
Funding
The authors did not receive financial support and sponsorship from individuals or organizations/institutions.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- Sachs J., Malaney P.. The economic and social burden of malaria. Nature. 2002;415(6872):680-5. View Article PubMed Google Scholar
- Trampuz A., Jereb M., Muzlovic I., Prabhu R.M.. Clinical review: severe malaria. Crit Care. 2003;7(4):315-23. View Article PubMed Google Scholar
- Svenson J.E., MacLean J.D., Gyorkos T.W., Keystone J.. Imported malaria. Clinical presentation and examination of symptomatic travelers. Arch Intern Med. 1995;155(8):861-8. View Article PubMed Google Scholar
- WHO. World Malaria Report 2016; 2017. Available from: http://www.who.int/malaria/publications/world-malaria-report-2016/en/. . ;:. Google Scholar
- Kakkilaya BS. Malaria Site: all about malaria. 2019. Available at https://www.malariasite.com/. . ;:. Google Scholar
- Dondorp A.M., Lee S.J., Faiz M.A., Mishra S., Price R., Tjitra E.. The relationship between age and the manifestations of and mortality associated with severe malaria. Clin Infect Dis. 2008;47(2):151-7. View Article PubMed Google Scholar
- von Seidlein L., Olaosebikan R., Hendriksen I.C., Lee S.J., Adedoyin O.T., Agbenyega T.. Predicting the clinical outcome of severe falciparum malaria in african children: findings from a large randomized trial. Clin Infect Dis. 2012;54(8):1080-90. View Article PubMed Google Scholar
- Plewes K., Turner G.D., Dondorp A.M.. Pathophysiology, clinical presentation, and treatment of coma and acute kidney injury complicating falciparum malaria. Curr Opin Infect Dis. 2018;31(1):69-77. View Article PubMed Google Scholar
- CDC. CDC yellow book-malaria. Oxford University Press. 2020. https://www.cdc.gov/malaria/about/disease.html. . ;:. Google Scholar
- Autino B., Corbett Y., Castelli F., Taramelli D.. Pathogenesis of malaria in tissues and blood. Mediterr J Hematol Infect Dis. 2012;4(1):e2012061. View Article PubMed Google Scholar
- Kyes S., Horrocks P., Newbold C.. Antigenic variation at the infected red cell surface in malaria. Annu Rev Microbiol. 2001;55(1):673-707. View Article PubMed Google Scholar
- Scherf A., Lopez-Rubio J.J., Riviere L.. Antigenic variation in Plasmodium falciparum. Annu Rev Microbiol. 2008;62(1):445-70. View Article PubMed Google Scholar
- Rowe J.A., Claessens A., Corrigan R.A., Arman M.. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Rev Mol Med. 2009;11:e16. View Article PubMed Google Scholar
- Kochar D.K., Shubhakaran Kumawat B.L., Kochar S.K., Halwai M., Makkar R.K.. Cerebral malaria in Indian adults: a prospective study of 441 patients from Bikaner, north-west India. J Assoc Physicians India. 2002;50:234-41. PubMed Google Scholar
- Kochar D.K., Saxena V., Singh N., Kochar S.K., Kumar S.V., Das A.. Plasmodium vivax malaria. Emerg Infect Dis. 2005;11(1):132-4. View Article PubMed Google Scholar
- Anstey N.M., Handojo T., Pain M.C., Kenangalem E., Tjitra E., Price R.N.. Lung injury in vivax malaria: pathophysiological evidence for pulmonary vascular sequestration and posttreatment alveolar-capillary inflammation. J Infect Dis. 2007;195(4):589-96. View Article PubMed Google Scholar
- Tjitra E., Anstey N.M., Sugiarto P., Warikar N., Kenangalem E., Karyana M.. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med. 2008;5(6):e128. View Article PubMed Google Scholar
- Anstey N.M., Russell B., Yeo T.W., Price R.N.. The pathophysiology of vivax malaria. Trends Parasitol. 2009;25(5):220-7. View Article PubMed Google Scholar
- Daneshvar C., Davis T.M., Cox-Singh J., Rafa'ee M.Z., Zakaria S.K., Divis P.C.. Clinical and laboratory features of human Plasmodium knowlesi infection. Clin Infect Dis. 2009;49(6):852-60. View Article PubMed Google Scholar
- Cox-Singh J., Hiu J., Lucas S.B., Divis P.C., Zulkarnaen M., Chandran P.. Severe malaria - a case of fatal Plasmodium knowlesi infection with post-mortem findings: a case report. Malar J. 2010;9(1):10. View Article PubMed Google Scholar
- Mackintosh C.L., Beeson J.G., Marsh K.. Clinical features and pathogenesis of severe malaria. Trends Parasitol. 2004;20(12):597-603. View Article PubMed Google Scholar
- Chakravorty S.J., Hughes K.R., Craig A.G.. Host response to cytoadherence in Plasmodium falciparum. Biochem Soc Trans. 2008;36(Pt 2):221-8. View Article PubMed Google Scholar
- Schofield L., Grau G.E.. Immunological processes in malaria pathogenesis. Nat Rev Immunol. 2005;5(9):722-35. View Article PubMed Google Scholar
- Clark I.A., Budd A.C., Alleva L.M., Cowden W.B.. Human malarial disease: a consequence of inflammatory cytokine release. Malar J. 2006;5(1):85. View Article PubMed Google Scholar
- Parroche P., Lauw F.N., Goutagny N., Latz E., Monks B.G., Visintin A.. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci USA. 2007;104(6):1919-24. View Article PubMed Google Scholar
- Schumann R.R.. Malarial fever: hemozoin is involved but Toll-free. Proc Natl Acad Sci USA. 2007;104(6):1743-4. View Article PubMed Google Scholar
- Awandare G.A., Ouma Y., Ouma C., Were T., Otieno R., Keller C.C.. Role of monocyte-acquired hemozoin in suppression of macrophage migration inhibitory factor in children with severe malarial anemia. Infect Immun. 2007;75(1):201-10. View Article PubMed Google Scholar
- Lamikanra A.A., Theron M., Kooij T.W., Roberts D.J.. Hemozoin (malarial pigment) directly promotes apoptosis of erythroid precursors. PLoS One. 2009;4(12):e8446. View Article PubMed Google Scholar
- Chen Q., Schlichtherle M., Wahlgren M.. Molecular aspects of severe malaria. Clin Microbiol Rev. 2000;13(3):439-50. View Article PubMed Google Scholar
- Naik R.S., Branch O.H., Woods A.S., Vijaykumar M., Perkins D.J., Nahlen B.L.. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J Exp Med. 2000;192(11):1563-76. View Article PubMed Google Scholar
- Udomsangpetch R., Pipitaporn B., Silamut K., Pinches R., Kyes S., Looareesuwan S.. Febrile temperatures induce cytoadherence of ring-stage Plasmodium falciparum-infected erythrocytes. Proc Natl Acad Sci USA. 2002;99(18):11825-9. View Article PubMed Google Scholar
- Omer F.M., de Souza J.B., Riley E.M.. Differential induction of TGF-beta regulates proinflammatory cytokine production and determines the outcome of lethal and nonlethal Plasmodium yoelii infections. J Immunol. 2003;171(10):5430-6. View Article PubMed Google Scholar
- van der Heyde H.C., Nolan J., Combes V., Gramaglia I., Grau G.E.. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22(11):503-8. View Article PubMed Google Scholar
- Poovassery J.S., Sarr D., Smith G., Nagy T., Moore J.M.. Malaria-induced murine pregnancy failure: distinct roles for IFN-γ and TNF. J Immunol. 2009;183(8):5342-9. View Article PubMed Google Scholar
- Faille D., Combes V., Mitchell A.J., Fontaine A., Juhan-Vague I., Alessi M.C.. Platelet microparticles: a new player in malaria parasite cytoadherence to human brain endothelium. FASEB J. 2009;23(10):3449-58. View Article PubMed Google Scholar
- Karunaweera N., Wanasekara D., Chandrasekharan V., Mendis K., Carter R.. Plasmodium vivax: paroxysm-associated lipids mediate leukocyte aggregation. Malar J. 2007;6(1):62. View Article PubMed Google Scholar
- Li I.W., Cheung L.C.. How not to miss a case of malaria in emergency department in malaria non-endemic areas? Practical approach and experiences in Hong Kong. Open J Emerg Med.. 2016;4(4):93-109. View Article Google Scholar
- Franke-Fayard B., Fonager J., Braks A., Khan S.M., Janse C.J.. Sequestration and tissue accumulation of human malaria parasites: can we learn anything from rodent models of malaria?. PLoS Pathog. 2010;6(9):e1001032. View Article PubMed Google Scholar
- Grau G.E., Craig A.G.. Cerebral malaria pathogenesis: revisiting parasite and host contributions. Future Microbiol. 2012;7(2):291-302. View Article PubMed Google Scholar
- Dondorp A.M., Pongponratn E., White N.J.. Reduced microcirculatory flow in severe falciparum malaria: pathophysiology and electron-microscopic pathology. Acta Trop. 2004;89(3):309-17. View Article PubMed Google Scholar
- Chakravorty S.J., Craig A.. The role of ICAM-1 in Plasmodium falciparum cytoadherence. Eur J Cell Biol. 2005;84(1):15-27. View Article PubMed Google Scholar
- Milner D.A.. Rethinking cerebral malaria pathology. Curr Opin Infect Dis. 2010;23(5):456-63. View Article PubMed Google Scholar
- Fried M., Domingo G.J., Gowda C.D., Mutabingwa T.K., Duffy P.E.. Plasmodium falciparum: chondroitin sulfate A is the major receptor for adhesion of parasitized erythrocytes in the placenta. Exp Parasitol. 2006;113(1):36-42. View Article PubMed Google Scholar
- Rogerson S.J., Mwapasa V., Meshnick S.R.. Malaria in pregnancy: linking immunity and pathogenesis to prevention. Am J Trop Med Hyg. 2007;77(6):14-22. View Article PubMed Google Scholar
- von Itzstein M., Plebanski M., Cooke B.M., Coppel R.L.. Hot, sweet and sticky: the glycobiology of Plasmodium falciparum. Trends Parasitol. 2008;24(5):210-8. View Article PubMed Google Scholar
- Yale S.H., Adlakha A., Sebo T.J., Ryu J.H.. Bronchiolitis obliterans organizing pneumonia caused by Plasmodium vivax malaria. Chest. 1993;104(4):1294-6. View Article PubMed Google Scholar
- Vogt A.M., Barragan A., Chen Q., Kironde F., Spillmann D., Wahlgren M.. Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the DBL1alpha domain of PfEMP1. Blood. 2003;101(6):2405-11. View Article PubMed Google Scholar
- Cockburn I.A., Mackinnon M.J., O'Donnell A., Allen S.J., Moulds J.M., Baisor M.. A human complement receptor 1 polymorphism that reduces Plasmodium falciparum rosetting confers protection against severe malaria. Proc Natl Acad Sci USA. 2004;101(1):272-7. View Article PubMed Google Scholar
- Horata N., Kalambaheti T., Craig A., Khusmith S.. Sequence variation of PfEMP1-DBLalpha in association with rosette formation in Plasmodium falciparum isolates causing severe and uncomplicated malaria. Malar J. 2009;8(1):184. View Article PubMed Google Scholar
- Rowe J.A., Handel I.G., Thera M.A., Deans A.M., Lyke K.E., Koné A.. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci USA. 2007;104(44):17471-6. View Article PubMed Google Scholar
- Cserti C.M., Dzik W.H.. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007;110(7):2250-8. View Article PubMed Google Scholar
- Doumbo O.K., Thera M.A., Koné A.K., Raza A., Tempest L.J., Lyke K.E.. High levels of Plasmodium falciparum rosetting in all clinical forms of severe malaria in African children. Am J Trop Med Hyg. 2009;81(6):987-93. View Article PubMed Google Scholar
- Uyoga S., Skorokhod O.A., Opiyo M., Orori E.N., Williams T.N., Arese P.. Transfer of 4-hydroxynonenal from parasitized to non-parasitized erythrocytes in rosettes. Proposed role in severe malaria anemia. Br J Haematol. 2012;157(1):116-24. View Article PubMed Google Scholar
- Cooke B.M., Mohandas N., Coppel R.L.. Malaria and the red blood cell membrane. Semin Hematol. 2004;41(2):173-88. View Article PubMed Google Scholar
- Nuchsongsin F., Chotivanich K., Charunwatthana P., Omodeo-Salè F., Taramelli D., Day N.P.. Effects of malaria heme products on red blood cell deformability. Am J Trop Med Hyg. 2007;77(4):617-22. View Article PubMed Google Scholar
- Park Y., Diez-Silva M., Popescu G., Lykotrafitis G., Choi W., Feld M.S.. Refractive index maps and membrane dynamics of human red blood cells parasitized by Plasmodium falciparum. Proc Natl Acad Sci USA. 2008;105(37):13730-5. View Article PubMed Google Scholar
- Handayani S., Chiu D.T., Tjitra E., Kuo J.S., Lampah D., Kenangalem E.. High deformability of Plasmodium vivax-infected red blood cells under microfluidic conditions. J Infect Dis. 2009;199(3):445-50. View Article PubMed Google Scholar
- Greenwood B.M., Fidock D.A., Kyle D.E., Kappe S.H., Alonso P.L., Collins F.H.. Malaria: progress, perils, and prospects for eradication. J Clin Invest. 2008;118(4):1266-76. View Article PubMed Google Scholar
- Nussenblatt V., Semba R.D.. Micronutrient malnutrition and the pathogenesis of malarial anemia. Acta Trop. 2002;82(3):321-37. View Article PubMed Google Scholar
- Ghosh K., Ghosh K.. Pathogenesis of anemia in malaria: a concise review. Parasitol Res. 2007;101(6):1463-9. View Article PubMed Google Scholar
- Haldar K., Mohandas N.. Malaria, erythrocytic infection, and anemia. Hematology (Am Soc Hematol Educ Program). 2009;1(1):87-93. View Article PubMed Google Scholar
- Skorokhod O.A., Caione L., Marrocco T., Migliardi G., Barrera V., Arese P.. Inhibition of erythropoiesis in malaria anemia: role of hemozoin and hemozoin-generated 4-hydroxynonenal. Blood. 2010;116(20):4328-37. View Article PubMed Google Scholar
- Buffet P.A., Safeukui I., Milon G., Mercereau-Puijalon O., David P.H.. Retention of erythrocytes in the spleen: a double-edged process in human malaria. Curr Opin Hematol. 2009;16(3):157-64. View Article PubMed Google Scholar
- Perkins D.J., Were T., Davenport G.C., Kempaiah P., Hittner J.B., Ong'echa J.M.. Severe malarial anemia: innate immunity and pathogenesis. Int J Biol Sci. 2011;7(9):1427-42. View Article PubMed Google Scholar
- McDevitt M.A., Xie J., Ganapathy-Kanniappan S., Griffith J., Liu A., McDonald C.. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203(5):1185-96. View Article PubMed Google Scholar
- Nweneka C.V., Doherty C.P., Cox S., Prentice A.. Iron delocalisation in the pathogenesis of malarial anaemia. Trans R Soc Trop Med Hyg. 2010;104(3):175-84. View Article PubMed Google Scholar
- de Mast Q., Syafruddin D., Keijmel S., Riekerink T.O., Deky O., Asih P.B.. Increased serum hepcidin and alterations in blood iron parameters associated with asymptomatic P. falciparum and P. vivax malaria. Haematologica. 2010;95(7):1068-74. View Article PubMed Google Scholar
- Maina R.N., Walsh D., Gaddy C., Hongo G., Waitumbi J., Otieno L.. Impact of Plasmodium falciparum infection on haematological parameters in children living in Western Kenya. Malar J. 2010;9(S3):4. View Article PubMed Google Scholar
- Kochar D.K., Das A., Kochar A., Middha S., Acharya J., Tanwar G.S.. Thrombocytopenia in Plasmodium falciparum, Plasmodium vivax and mixed infection malaria: a study from Bikaner (Northwestern India). Platelets. 2010;21(8):623-7. View Article PubMed Google Scholar
- Leowattana W., Tangpukdee N., Thar S.K., Nakasiri S., Srivilairit S., Kano S.. Changes in platelet count in uncomplicated and severe falciparum malaria. Southeast Asian J Trop Med Public Health. 2010;41(5):1035-41. PubMed Google Scholar
- Tan S.O., McGready R., Zwang J., Pimanpanarak M., Sriprawat K., Thwai K.L.. Thrombocytopaenia in pregnant women with malaria on the Thai-Burmese border. Malar J. 2008;7(1):209. View Article PubMed Google Scholar
- Lacerda M.V., Mourão M.P., Coelho H.C., Santos J.B.. Thrombocytopenia in malaria: who cares?. Mem Inst Oswaldo Cruz. 2011;106:52-63. View Article PubMed Google Scholar
- de Mast Q., Groot E., Asih P.B., Syafruddin D., Oosting M., Sebastian S.. ADAMTS13 deficiency with elevated levels of ultra-large and active von Willebrand factor in P. falciparum and P. vivax malaria. Am J Trop Med Hyg. 2009;80(3):492-8. View Article PubMed Google Scholar
- Association American Medical. Acute respiratory distress syndrome. . 2012;:. Google Scholar
- Mohan A., Sharma S.K., Bollineni S.. Acute lung injury and acute respiratory distress syndrome in malaria. J Vector Borne Dis. 2008;45(3):179-93. PubMed Google Scholar
- Van den Steen P.E., Deroost K., Deckers J., Van Herck E., Struyf S., Opdenakker G.. Pathogenesis of malaria-associated acute respiratory distress syndrome. Trends Parasitol. 2013;29(7):346-58. View Article PubMed Google Scholar
- Anstey N.M., Jacups S.P., Cain T., Pearson T., Ziesing P.J., Fisher D.A.. Pulmonary manifestations of uncomplicated falciparum and vivax malaria: cough, small airways obstruction, impaired gas transfer, and increased pulmonary phagocytic activity. J Infect Dis. 2002;185(9):1326-34. View Article PubMed Google Scholar
- Kingston H., Turner G., Day N., Hommel M., Kremsner P.. Lung involvement in malaria. . 2015;:. View Article Google Scholar
- Grommes J., Soehnlein O.. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17(3-4):293-307. View Article PubMed Google Scholar
- Aitken E.H., Negri E.M., Barboza R., Lima M.R., Álvarez J.M., Marinho C.R.. Ultrastructure of the lung in a murine model of malaria-associated acute lung injury/acute respiratory distress syndrome. Malar J. 2014;13(1):230. View Article PubMed Google Scholar
- Ortolan L.S., Sercundes M.K., Barboza R., Debone D., Murillo O., Hagen S.C.F.. Predictive Criteria to Study the Pathogenesis of Malaria-Associated ALI/ARDS in Mice. Mediators of Inflammation. 2014;2014:872464. View Article Google Scholar
- Ampawong S., Chaisri U., Viriyavejakul P., Prapansilp P., Grau G.E., Turner G.D.. A potential role for interleukin-33 and γ-epithelium sodium channel in the pathogenesis of human malaria associated lung injury. Malar J. 2015;14(1):389. View Article PubMed Google Scholar
- de Azevedo-Quintanilha I.G., Vieira-de-Abreu A., Ferreira A.C., Nascimento D.O., Siqueira A.M., Campbell R.A.. Integrin αDβ2 (CD11d/CD18) mediates experimental malaria-associated acute respiratory distress syndrome (MA-ARDS). Malar J. 2016;15(1):393. View Article PubMed Google Scholar
- Pham T.T., Verheijen M., Vandermosten L., Deroost K., Knoops S., Van den Eynde K.. Pathogenic CD8+ T cells cause increased levels of VEGF-A in experimental malaria-associated acute respiratory distress syndrome, but therapeutic VEGFR inhibition is not effective. Front Cell Infect Microbiol. 2017;7:416. View Article PubMed Google Scholar
- Epiphanio S., Campos M.G., Pamplona A., Carapau D., Pena A.C., Ataíde R.. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog. 2010;6(5):e1000916. View Article PubMed Google Scholar
- Maknitikul S., Luplertlop N., Grau G.E., Ampawong S.. Dysregulation of pulmonary endothelial protein C receptor and thrombomodulin in severe falciparum malaria-associated ARDS relevant to hemozoin. PLoS One. 2017;12(7):e0181674. View Article PubMed Google Scholar
- Oberli A., Zurbrügg L., Rusch S., Brand F., Butler M.E., Day J.L.. Plasmodium falciparum Plasmodium helical interspersed subtelomeric proteins contribute to cytoadherence and anchor P. falciparum erythrocyte membrane protein 1 to the host cell cytoskeleton. Cell Microbiol. 2016;18(10):1415-28. View Article PubMed Google Scholar
- Wassmer S.C., Grau G.E., Grau R.. Severe malaria: what's new on the pathogenesis front?. Int J Parasitol. 2017;47(2-3):145-52. View Article PubMed Google Scholar
- Deroost K., Tyberghein A., Lays N., Noppen S., Schwarzer E., Vanstreels E.. Hemozoin induces lung inflammation and correlates with malaria-associated acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2013;48(5):589-600. View Article PubMed Google Scholar
- Pereira M.L., Ortolan L.S., Sercundes M.K., Debone D., Murillo O., Lima F.A.. Association of heme oxygenase 1 with lung protection in malaria-associated ALI/ARDS. . 2016;:4158698. Google Scholar
- Pasternak N.D., Dzikowski R.. PfEMP1: an antigen that plays a key role in the pathogenicity and immune evasion of the malaria parasite Plasmodium falciparum. Int J Biochem Cell Biol. 2009;41(7):1463-6. View Article PubMed Google Scholar
- Turner L., Lavstsen T., Berger S.S., Wang C.W., Petersen J.E., Avril M.. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498(7455):502-5. View Article PubMed Google Scholar
- Sercundes M.K., Ortolan L.S., Debone D., Soeiro-Pereira P.V., Gomes E., Aitken E.H.. Targeting neutrophils to prevent malaria-associated acute lung injury/acute respiratory distress syndrome in mice. PLoS Pathog. 2016;12(12):e1006054. View Article PubMed Google Scholar
- Boeltz S., Muñoz L.E., Fuchs T.A., Herrmann M.. Neutrophil extracellular traps open the Pandora's box in severe malaria. Front Immunol. 2017;8:874. View Article PubMed Google Scholar
- Warrell D.A., Looareesuwan S., Warrell M.J., Kasemsarn P., Intaraprasert R., Bunnag D.. Dexamethasone proves deleterious in cerebral malaria. A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306(6):313-9. View Article PubMed Google Scholar
- Brewster D.R., Kwiatkowski D., White N.J.. Neurological sequelae of cerebral malaria in children. Lancet. 1990;336(8722):1039-43. View Article PubMed Google Scholar
- Beare N.A., Taylor T.E., Harding S.P., Lewallen S., Molyneux M.E.. Malarial retinopathy: a newly established diagnostic sign in severe malaria. Am J Trop Med Hyg. 2006;75(5):790-7. View Article PubMed Google Scholar
- Maude R.J., Hassan M.U., Beare N.A.. Severe retinal whitening in an adult with cerebral malaria. Am J Trop Med Hyg. 2009;80(6):881. View Article PubMed Google Scholar
- Rupani A.B., Amarapurkar A.D.. Hepatic changes in fatal malaria: an emerging problem. Ann Trop Med Parasitol. 2009;103(2):119-27. View Article PubMed Google Scholar
- Whitten R., Milner D.A., Yeh M.M., Kamiza S., Molyneux M.E., Taylor T.E.. Liver pathology in Malawian children with fatal encephalopathy. Hum Pathol. 2011;42(9):1230-9. View Article PubMed Google Scholar
- Prommano O., Chaisri U., Turner G.D., Wilairatana P., Ferguson D.J., Viriyavejakul P.. A quantitative ultrastructural study of the liver and the spleen in fatal falciparum malaria. Southeast Asian J Trop Med Public Health. 2005;36(6):1359-70. PubMed Google Scholar
- Guha M., Kumar S., Choubey V., Maity P., Bandyopadhyay U., Guha M.. Apoptosis in liver during malaria: role of oxidative stress and implication of mitochondrial pathway. FASEB J. 2006;20(8):1224-6. View Article PubMed Google Scholar
- Dey S., Guha M., Alam A., Goyal M., Bindu S., Pal C.. Malarial infection develops mitochondrial pathology and mitochondrial oxidative stress to promote hepatocyte apoptosis. Free Radic Biol Med. 2009;46(2):271-81. View Article PubMed Google Scholar
- Klotz C., Frevert U.. Plasmodium yoelii sporozoites modulate cytokine profile and induce apoptosis in murine Kupffer cells. Int J Parasitol. 2008;38(14):1639-50. View Article PubMed Google Scholar
- Thompson C.B.. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267(5203):1456-62. View Article PubMed Google Scholar
- Thornberry N.A.. Caspases: key mediators of apoptosis. Chem Biol. 1998;5(5):97-103. View Article PubMed Google Scholar
- Shishodia S., Aggarwal B.B.. Nuclear factor-kappaB: a friend or a foe in cancer?. Biochem Pharmacol. 2004;68(6):1071-80. View Article PubMed Google Scholar
- Punsawad C., Maneerat Y., Chaisri U., Nantavisai K., Viriyavejakul P.. Nuclear factor kappa B modulates apoptosis in the brain endothelial cells and intravascular leukocytes of fatal cerebral malaria. Malar J. 2013;12(1):260-10. View Article PubMed Google Scholar
- Kochar D.P., Sirohi P., Kochar A.. Acute hepatitis in malaria. J Gastroenterol Hepatol. 2006;2:36-8. PubMed Google Scholar
- Baheti R., Laddha P., Gehlot R.S.. Liver involvement in falciparum malaria: a histo-pathological analysis. JIACM. 2003;4:34-8. Google Scholar
- WHO World Malaria Report 2000. . 2000;:. Google Scholar
- Kochar D.K., Agarwal P., Kochar S.K., Jain R., Rawat N., Pokharna R.K.. Hepatocyte dysfunction and hepatic encephalopathy in Plasmodium falciparum malaria. QJM. 2003;96(7):505-12. View Article PubMed Google Scholar
- Bhalla A., Suri V., Singh V.. Malarial hepatopathy. J Postgrad Med. 2006;52(4):315-20. PubMed Google Scholar
- Iwalokun B.A., Bamiro S.B., Ogunledun A.. Levels and interactions of plasma xanthine oxidase, catalase and liver function parameters in Nigerian children with Plasmodium falciparum infection. Acta Pathol Microbiol Scand Suppl. 2006;114(12):842-50. View Article PubMed Google Scholar
- Elsheikha H.M., Sheashaa H.A.. Epidemiology, pathophysiology, management and outcome of renal dysfunction associated with plasmodia infection. Parasitol Res. 2007;101(5):1183-90. View Article PubMed Google Scholar
- Barsoum R.S.. Malarial acute renal failure. J Am Soc Nephrol. 2000;11(11):2147-54. View Article PubMed Google Scholar
- Naqvi R.. Plasmodium Vivax causing acute kidney injury: A foe less addressed. Pak J Med Sci. 2015;31(6):1472-5. PubMed Google Scholar
- Gomes A.P., Vitorino R.R., Costa A.P., Mendonça E.G., Oliveira M.G., Siqueira-Batista R.. Malária grave por Plasmodium falciparum. Rev Bras Ter Intensiva. 2011;23(3):358-69. View Article PubMed Google Scholar
- Kute V.B., Trivedi H.L., Vanikar A.V., Shah P.R., Gumber M.R., Patel H.V.. Plasmodium vivax malaria-associated acute kidney injury, India, 2010-2011. Emerg Infect Dis. 2012;18(5):842-5. View Article PubMed Google Scholar
- Saravu K., Rishikesh K., Parikh C.R.. Risk factors and outcomes stratified by severity of acute kidney injury in malaria. PLoS One. 2014;9(3):e90419. View Article PubMed Google Scholar
- Mohapatra M.K., Behera A.K., Karua P.C., Bariha P.K., Rath A., Aggrawal K.C.. Urinary bile casts in bile cast nephropathy secondary to severe falciparum malaria. Clin Kidney J. 2016;9(4):644-8. View Article PubMed Google Scholar
- Win K.K., Thanachartwet V., Wattanagoon Y., Jerraksuwan S., Ruangweerayut R., Desakorn V.. Factors associated with acute renal failure in adults with severe falciparum malaria. Southeast Asian J Trop Med Public Health. 2012;43(5):1071-9. PubMed Google Scholar
- Siriwardhana E.A., Perera P.A., Sivakanesan R., Abeysekara T., Nugegoda D.B., Jayaweera J.A.. Dehydration and malaria augment the risk of developing chronic kidney disease in Sri Lanka. Indian J Nephrol. 2015;25(3):146-51. View Article PubMed Google Scholar
- Zakama A.K., Gaw S.L.. Malaria in pregnancy: what the obstetric provider in non-endemic areas needs to know. Obstet Gynecol Surv. 2019;74(9):546-56. View Article PubMed Google Scholar
- Lawford H.L., Lee A.C., Kumar S., Liley H.G., Bora S.. Establishing a conceptual framework of the impact of placental malaria on infant neurodevelopment. Int J Infect Dis. 2019;84:54-65. View Article PubMed Google Scholar
- Feleke D.G., Adamu A., Gebreweld A., Tesfaye M., Demisiss W., Molla G.. Asymptomatic malaria infection among pregnant women attending antenatal care in malaria endemic areas of North-Shoa, Ethiopia: a cross-sectional study. Malar J. 2020;19(1):67. View Article PubMed Google Scholar
- Menendez C., Ordi J., Ismail M.R., Ventura P.J., Aponte J.J., Kahigwa E.. The impact of placental malaria on gestational age and birth weight. J Infect Dis. 2000;181(5):1740-5. View Article PubMed Google Scholar
- WHO World malaria report 2017. . 2017;:. Google Scholar
- Clark R.L.. Genesis of placental sequestration in malaria and possible targets for drugs for placental malaria. Birth Defects Res. 2019;111(10):569-83. View Article PubMed Google Scholar
- McDonald C.R., Tran V., Kain K.C.. Complement activation in placental malaria. Front Microbiol. 2015;6:1460. View Article PubMed Google Scholar
- Pehrson C., Salanti A., Theander T.G., Nielsen M.A.. Pre-clinical and clinical development of the first placental malaria vaccine. Expert Rev Vaccines. 2017;16(6):613-24. View Article PubMed Google Scholar
- Reis A.S., Barboza R., Murillo O., Barateiro A., Peixoto E.P., Lima F.A.. Inflammasome activation and IL-1 signaling during placental malaria induce poor pregnancy outcomes. Sci Adv. 2020;6(10):. View Article PubMed Google Scholar
- Lima F.A., Barateiro A., Dombrowski J.G., de Souza R.M., Costa D.S., Murillo O.. Plasmodium falciparum infection dysregulates placental autophagy. PLoS One. 2019;14(12):e0226117. View Article PubMed Google Scholar
- Dimasuay K.G., Gong L., Rosario F., McBryde E., Spelman T., Glazier J.. Impaired placental autophagy in placental malaria. PLoS One. 2017;12(11):e0187291. View Article PubMed Google Scholar
- Megnekou R., Tenou S., Bigoga J.D., Djontu J.C., Medou F.M., Lissom A.. Placental malaria and modulation of immune and hormonal responses in Cameroonian women. Acta Trop. 2015;147:23-30. View Article PubMed Google Scholar
- Sharma L., Shukla G.. Placental malaria: A new insight into the pathophysiology. Front Med (Lausanne). 2017;4:117. View Article PubMed Google Scholar
- Obiri D., Erskine I.J., Oduro D., Kusi K.A., Amponsah J., Gyan B.A.. Histopathological lesions and exposure to Plasmodium falciparum infections in the placenta increases the risk of preeclampsia among pregnant women. Sci Rep. 2020;10(1):8280. View Article PubMed Google Scholar
- Odorizzi P.M., Feeney M.E.. Impact of in utero exposure to malaria on fetal T cell immunity. Trends Mol Med. 2016;22(10):877-88. View Article PubMed Google Scholar
- Kidima W.B.. Syncytiotrophoblast functions and fetal growth restriction during placental malaria: updates and implication for future interventions. BioMed Res Int. 2015;2015:451735. View Article PubMed Google Scholar
- Kapisi J., Kakuru A., Jagannathan P., Muhindo M.K., Natureeba P., Awori P.. Relationships between infection with Plasmodium falciparum during pregnancy, measures of placental malaria, and adverse birth outcomes. Malar J. 2017;16(1):400. View Article PubMed Google Scholar
- Lybbert J., Gullingsrud J., Chesnokov O., Turyakira E., Dhorda M., Guerin P.J.. Abundance of megalin and Dab2 is reduced in syncytiotrophoblast during placental malaria, which may contribute to low birth weight. Sci Rep. 2016;6(1):24508. View Article PubMed Google Scholar
- Oktavianthi S., Fauzi M., Trianty L., Trimarsanto H., Bowolaksono A., Noviyanti R.. Placental mitochondrial DNA copy number is associated with reduced birth weight in women with placental malaria. Placenta. 2019;80:1-3. View Article PubMed Google Scholar
- Moeller S.L., Nyengaard J.R., Larsen L.G., Nielsen K., Bygbjerg I.C., Msemo O.A.. Malaria in early pregnancy and the development of the placental vasculature. J Infect Dis. 2019;220(9):1425-34. View Article PubMed Google Scholar
- Elphinstone R.E., Weckman A.M., McDonald C.R., Tran V., Zhong K., Madanitsa M.. Early malaria infection, dysregulation of angiogenesis, metabolism and inflammation across pregnancy, and risk of preterm birth in Malawi: A cohort study. PLoS Med. 2019;16(10):e1002914. View Article PubMed Google Scholar
- Steketee R.W., Wirima J.J., Bloland P.B., Chilima B., Mermin J.H., Chitsulo L.. Impairment of a pregnant woman's acquired ability to limit Plasmodium falciparum by infection with human immunodeficiency virus type-1. Am J Trop Med Hyg. 1996;55(1):42-9. View Article PubMed Google Scholar
- Natureeba P., Ades V., Luwedde F., Mwesigwa J., Plenty A., Okong P.. Lopinavir/ritonavir-based antiretroviral treatment (ART) versus efavirenz-based ART for the prevention of malaria among HIV-infected pregnant women. J Infect Dis. 2014;210(12):1938-45. View Article PubMed Google Scholar
- Boudová S., Divala T., Mungwira R., Mawindo P., Tomoka T., Laufer M.K.. Placental but not peripheral Plasmodium falciparum infection during pregnancy is associated with increased risk of malaria in infancy. J Infect Dis. 2017;216(6):732-5. View Article PubMed Google Scholar
- Tassi Y.S., Fouda G.G., Sama G., Ngu J.B., Leke R.G., Taylor D.W.. Increased susceptibility to Plasmodium falciparum in infants is associated with low, not high, placental malaria parasitaemia. Sci Rep. 2018;8:1-10. Google Scholar
- Hangi M., Achan J., Saruti A., Quinlan J., Idro R.. Congenital malaria in newborns presented at Tororo General Hospital in Uganda: A cross-sectional study. Am J Trop Med Hyg. 2019;100(5):1158-63. View Article PubMed Google Scholar
- Adam I., Elhassan E.M., Haggaz A.E., Ali A.A., Adam G.K.. A perspective of the epidemiology of malaria and anaemia and their impact on maternal and perinatal outcomes in Sudan. J Infect Dev Ctries. 2011;5(2):83-7. View Article PubMed Google Scholar
- Rogerson S.J., Desai M., Mayor A., Sicuri E., Taylor S.M., van Eijk A.M.. Burden, pathology, and costs of malaria in pregnancy: new developments for an old problem. Lancet Infect Dis. 2018;18(4):e107-18. View Article PubMed Google Scholar
- Rogerson S.J., Pollina E., Getachew A., Tadesse E., Lema V.M., Molyneux M.E.. Placental monocyte infiltrates in response to Plasmodium falciparum malaria infection and their association with adverse pregnancy outcomes. Am J Trop Med Hyg. 2003;68(1):115-9. View Article PubMed Google Scholar
- Poovassery J., Moore J.M.. Murine malaria infection induces fetal loss associated with accumulation of Plasmodium chabaudi AS-infected erythrocytes in the placenta. Infect Immun. 2006;74(5):2839-48. View Article PubMed Google Scholar
- Davison B.B., Cogswell F.B., Baskin G.B., Falkenstein K.P., Henson E.W., Krogstad D.J.. Placental changes associated with fetal outcome in the Plasmodium coatneyi/rhesus monkey model of malaria in pregnancy. Am J Trop Med Hyg. 2000;63(3-4):158-73. View Article PubMed Google Scholar
- Ismail M.R., Ordi J., Menendez C., Ventura P.J., Aponte J.J., Kahigwa E.. Placental pathology in malaria: a histological, immunohistochemical, and quantitative study. Hum Pathol. 2000;31(1):85-93. View Article PubMed Google Scholar
- Saba N., Sultana A., Mahsud I.. Outcome and complications of malaria in pregnancy. Gomal J Med Sci.. 2008;6:98-101. Google Scholar
- Desai M., ter Kuile F.O., Nosten F., McGready R., Asamoa K., Brabin B.. Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis. 2007;7(2):93-104. View Article PubMed Google Scholar




